

Abstract
Introduction: Classical Hodgkin Lymphoma (CHL) is responsive to Programmed Death 1 inhibition (PD1i). Recognition that 9p24 amplifications promote PDL1 upregulation in lymphoma cells provides insight into CHL's particular sensitivity to PD1 inhibitors, but their mechanism of action remains unclear. Although it is widely believed that PD1i act by reversing CD8 T cell exhaustion, neither CD8 infiltration, PD1 expression nor lymphoma MHC class 1 (MHC1) expression predict PD1i response in CHL. Additionally, while most CHL cells adopt CD8 evasion mechanisms such as MHC1 loss, MHC2 expression is frequently retained suggesting a vulnerability to CD4-mediated cytotoxicity. CD4 cells dominate the CHL microenvironment and recent data demonstrates that MHC2 alongside PDL1 expression on CHL cells predict PD1i response suggesting an important role for CD4 cells in PD1i action.
Aim: To evaluate the impact of the PDL1 axis and MHC2 expression on the CD4 compartment in CHL and their implications for PD1i activity.
Methods: We evaluated 146 CHL patients by immunohistochemistry (IHC), with comparison to 24 reactive lymph node (RLN) controls. We designed a novel spatial analysis method utilising intra-tumor cell heterogeneity to test the influence of lymphoma PDL1 expression upon the T cell infiltrate. By combining IHC dual staining with stripping and reprobing we stained sections for CD30/PDL1/CD3 and serial sections on either side for GATA3/CD8, FoxP3/CD8, RORgT/CD8 and Tbet/CD8. Images were virtually overlaid and spatial analysis was performed using Visiopharm software. Distances from T cell subtypes were measured to either CD30+PDL1- CHL cells, CD30+PDL1+ CHL cells or CD30-PDL1+ microenvironmental cells. Skewing was tested statistically by comparison to parent cell distribution (e.g. CD8+ to CD3+). Spatial analysis was performed in 10 blindly selected cases.
Results/Discussion: Despite high PDL1 expression our data showed limited evidence for an exhausted PD1+CD8 population; CD8 tumor-infiltrating lymphocytes (TIL) were frequent, but PD1+TIL were rare (median 33% vs 0.08% nucleated cells respectively). No correlation was seen between PD1+TIL and CD8+TIL or PDL1. Stripping and reprobing of PD1+ IHC sections demonstrated that PD1+ cells were CD8 negative. This led us to consider alternative mechanisms of action for PD1i.
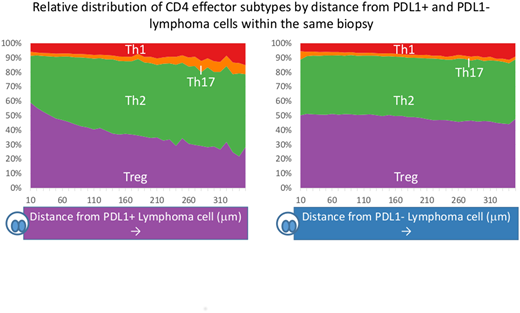
Since PD1i has a profound impact on naïve CD4 differentiation and influences the type of effector generated, we next quantified CD4 effector subtypes. We found that Tbet+TIL (Th1, antitumor) and FoxP3+TIL (Treg, protumor) were enriched in CHL, whereas RORgT+TIL (Th17, antitumor) were suppressed compared to RLN (p=<0.0001, 0.0004 and <0.0001 respectively). No significant difference was seen for GATA3+TIL (Th2, protumor). We further evaluated Treg and Th17 due to their shared developmental pathway which is known to be influenced by PDL1. FoxP3:Th17 ratios were heavily skewed towards Treg in CHL compared to RLN (p=<0.0001). Spatial analysis demonstrated enrichment of FoxP3+TIL and a paucity of RORgT+TIL in proximity to CD30+ lymphoma cells and with greater enrichment around PDL1+ lymphoma cells than PDL1- lymphoma cells. This supports a hypothesis of skewing of the Treg/Th17 axis by the lymphoma cell by the PD1-PDL1 axis. Finally, we evaluated the relationship between effector subtypes and CHL MHC2 expression across our cohort and found a positive association between FoxP3+TIL numbers and lymphoma cell MHC2 expression (p=0.0014).
Conclusion: The skewing of Th17/Treg via the PD1-PDL1 axis and positive association between Treg numbers and lymphoma MHC2 expression are consistent with CHL cells promoting Treg differentiation. This is supported in the literature by reports of MHC2-dependent induction of Treg by CHL cell lines. Paradoxically, Treg are associated with improved response to therapy despite evidence that they are induced by CHL cells. It is therefore hypothesised that Treg directly suppress lymphoma cell expansion. However, these data support our alternative hypothesis that PDL1 and MHC2 expression contribute to CHL cells establishing a Treg-rich and Th17-poor immunosuppressive microenvironment. Intervention with PD1i therapy or chemotherapy causing immunogenic cell death then shifts the infiltrate to a less suppressive environment leaving MHC2+ cells vulnerable to CD4-mediated cytotoxicity. We are currently modelling this in vitro.
Gribben:Abbvie: Honoraria; Medical Research Council: Research Funding; Novartis: Honoraria; Cancer Research UK: Research Funding; NIH: Research Funding; Acerta Pharma: Honoraria, Research Funding; Pharmacyclics: Honoraria; Celgene: Consultancy, Honoraria, Research Funding; Roche: Honoraria; TG Therapeutics: Honoraria; Janssen: Honoraria, Research Funding; Unum: Equity Ownership; Wellcome Trust: Research Funding; Kite: Honoraria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal